Resolving plant–fungal symbioses through multi-scale transcriptomics
The molecular mechanisms governing interactions between plants and their rhizosphere microbiomes remain poorly understood. Previously, using Populus as a model system, we helped conduct a large metatranscriptomic survey of natural aspen soil microbiomes, identified the cryptic root endophyte Hyaloscypha finlandica as the only core symbiont active across all sites and seasons, and revealed guild-specific seasonal shifts in carbon and nitrogen metabolism consistent with a transcriptomic phenology of fungal symbionts.
At the University of Illinois, we are establishing a Populus common garden to track how rhizosphere fungal communities and their gene expression programs shift across seasons and in response to environmental stress. Paired with our single-root-tip RNA-seq workflow, which recovers total RNA from individual colonized tips to resolve tri-partite host–fungal–bacterial interactions, and spatial transcriptomics in axenic microcosms, this work will generate transcriptomic atlases that pinpoint the enzyme, transporter, and signaling pathways underpinning nutrient exchange, colonization, and competition.
Collaborators: PMI Consortium (Oak Ridge National Lab).
How do fungal symbiotic functions evolve across the landscape and through time?
Which fungal traits matter for forest productivity, where do they occur, and why do they change across space and time? To answer this, we connect population genomics, comparative genomics, and in situ transcriptomics to move beyond community composition toward a mechanistic understanding of how symbiotic functions arise and shift across environments and evolutionary time. To this end we are actively developing substantial genomic resources for multiple charismatic soil fungi.
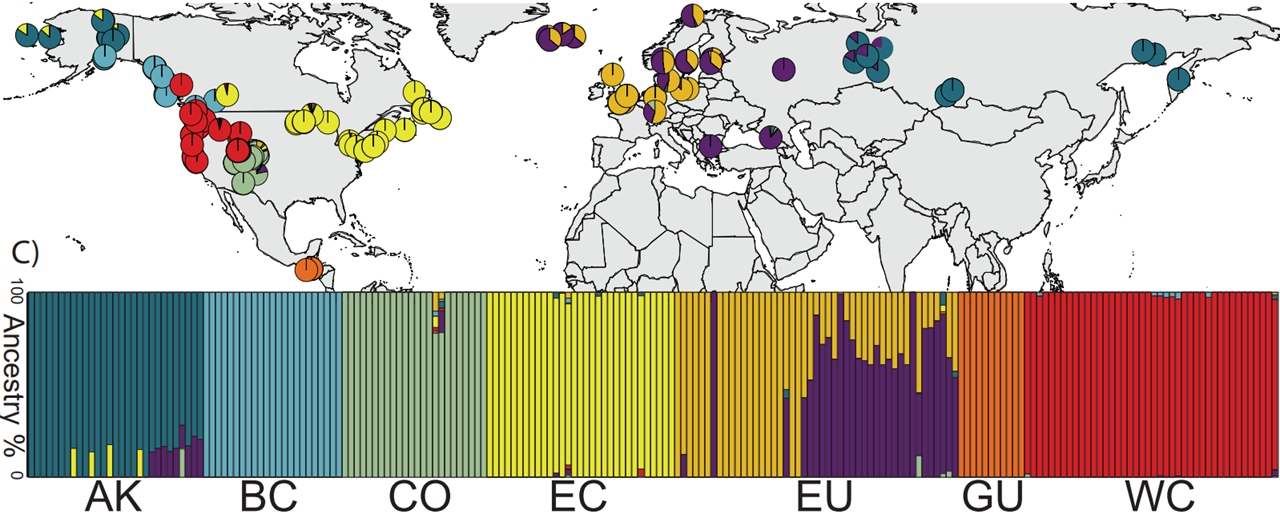
At the population scale, the porcini mushroom (Boletus edulis) serves as a model for trait evolution within and among populations of an essential forest symbiont. Since 2017 we have assembled one of the largest population-genomic datasets for any symbiotic fungus, 295 genomes across 16 countries and four continents, including 24 reference-quality long-read assemblies (one telomere-to-telomere). These genomes reveal cryptic, geographically structured lineages, gene flow among some but not all lineages, and decomposition repertoires that track local environment. Going forward, we will use these resources to dissect local adaptation, characterize hybridization where lineages meet in sympatry, and resolve how distinct ecological niches emerge within a single nominal species.
At the genus scale, Laccaria lets us ask how the symbiotic toolkit is gained, lost, and repurposed over deeper time. From 120 new genomes we built a genus-wide "super-pangenome" revealing repeated niche transitions, genome rearrangements, and lineage-specific gene-family expansions, consistent with a two-speed genome whose accessory compartment carries much of the symbiotic hardware. Even canonical effectors essential for host colonization show dynamic histories.
Integrating these resources with metatranscriptomes from colonized roots, we connect genomic variation to realized function in situ — building trait maps that link landscape heterogeneity to functional diversity and symbiotic performance.
Collaborators: Hoffman Lab (Bielefeld University), INRAE (Nance, France).

How has the fungal symbiosis toolkit diversified over deep time?
Ectomycorrhizal fungal diversity reflects hundreds of millions of years of evolutionary divergence, niche transitions, and genomic innovation. Focusing on the porcini family Boletaceae (>2,000 species), we combine dense herbarium and tropical field sampling with genome-scale phylogenetics to determine when major radiations occurred and how they intersect with geological and biogeographic history.
The lab generates comparative genomic resources across the family, tracking evolution in core symbiosis genes and genome architecture. Pairing these data with ancestral host reconstructions and available transcriptomes lets us test which components of the fungal "symbiosis toolkit" are conserved, and which diversify with new hosts or environments, revealing how deep-time processes enabled the ecological differentiation we observe in forest soils today.